Why MedTech Teams Can No Longer Afford Fragmented Development
This article explores why fragmented development models are no longer sustainable in MedTech, as connected devices, continuous software updates, and evolving regulations demand reproducible, compliance-ready development workflows across the entire device lifecycle.

21 May, 2026. 6 minutes read
Medical device development has long been organized around hardware stability and infrequent software change. With minimal connectivity, compliance was handled at the end of the cycle through documentation, validation, and submission preparation. That sequencing aligned with the constraints of those systems.
As medical device connectivity has expanded, regulatory expectations extend across the full software lifecycle. IEC 62304 governs development, maintenance, and change management. FDA cybersecurity guidance requires documented threat modeling, secure update mechanisms, and post-market monitoring. For connected devices operating across hospital networks, IEC 62443 adds network-level cybersecurity requirements that sit alongside these obligations. The EU Cyber Resilience Act (CRA) mandates early warning notifications within 24 hours of an actively exploited vulnerability and full reporting within 72 hours. All apply throughout the active life of the device.
Medical devices increasingly operate as connected, software-defined systems. Over-the-Air (OTA) update capability, network interfaces, and cloud connectivity introduce continuous exposure. Each update cycle becomes a compliance event where the software must be validated, the build reproducible, and the evidence organized. Fragmented toolchains and disconnected workflows introduce gaps in traceability, validation, and response readiness, increasing both cost and operational risk.
From Stable Devices to Continuous Obligations
Certification as a terminal event does not support connected device development. Devices that receive software updates after deployment require each update to pass through a documented development, validation, and release process. Patches issued in response to vulnerabilities must meet the same traceability and quality requirements as the original release and must be delivered within defined regulatory timelines.
Lifecycle responsibility extends beyond initial certification. Medical devices commonly remain in active use for 10 to 15 years, and software support obligations persist across that period. Software updates, patches, and security fixes must be delivered and validated repeatedly under controlled conditions. Compliance now includes response time, not only validation.
CRA reporting timelines illustrate the operational pressure this creates. A 24-hour early warning window for an actively exploited vulnerability requires immediate identification of the affected software component, including its specific version and associated SBOM entry, supported by traceability and an operational response workflow. Teams that assemble compliance evidence manually across fragmented systems cannot meet this requirement consistently, as evidence must be reconstructed across multiple tools under strict time constraints.
The Cost of Fragmented Development in MedTech
Fragmentation in MedTech development accumulates across toolchain configuration, evidence collection, and workflow integration. The resulting costs scale with both system complexity and regulatory pressure.
Most teams rely on separate tools for compilation, static analysis, testing, and security assessment. Each tool introduces its own configuration, versioning model, and output format. Manual integration between these systems creates ambiguity around which tool version produced a result, under which configuration, and against which source state. Traceability becomes difficult to establish during audits.
Each toolchain component in a regulated environment requires validation. Teams must demonstrate that tools behave correctly under defined conditions of use. When tools are sourced and integrated independently, this validation effort increases with the number of components and must be repeated across product lines and releases.
As software complexity grows, test coverage expands, analysis scope increases, and the number of configurations requiring validation rises. Safety and security workflows often operate in parallel without integration, producing separate evidence trails that must be reconciled during submissions. This accumulates as software lifecycle debt, where each release inherits unresolved complexity from previous development cycles. The impact becomes visible during audits, security incidents, and post-market updates.
The business consequences are direct. Delayed regulatory submissions affect revenue timelines. Rework driven by late-cycle defect discovery reduces engineering capacity. Compliance gaps identified during audits extend validation cycles and introduce additional remediation effort at the portfolio level.
What a Compliance-Ready Development Model Looks Like
MedTech teams adapting to current regulatory requirements are aligning their development systems around three operational shifts that integrate validation, execution, and compliance into daily development.
Shift 1: Verification Moves into Daily Development
Static analysis, traceability, and risk assessment are integrated into the developer workflow. Coding standards including MISRA C/C++, CERT C/C++, and CWE guidelines are enforced at commit level, ensuring that issues are identified in context and addressed immediately. Early detection reduces downstream validation effort and limits the propagation of defects into later stages of development. Compliance evidence is generated continuously as part of normal development activity.
Shift 2: Reproducible, Deterministic Pipelines
Containerized build environments define a consistent execution context across developer machines and CI infrastructure. Toolchains, dependencies, and configurations are versioned and controlled. Build, test, and analysis outputs are linked to specific source states and stored as structured artifacts. Each pipeline execution produces repeatable results that can be audited directly from recorded artifacts. CI/CD infrastructure enforces repeatability across environments and over time.
Shift 3: Toolchains Become Part of the Compliance Case
Tool qualification contributes directly to the compliance framework. Certified toolchains reduce the need for internal validation of tool behavior and limit the scope of audit effort. A pre-qualified toolchain defines a stable boundary for build, analysis, and validation. Unified development platforms that support multiple architectures reduce the number of separate qualification cases required across product lines. Integrated toolchains also enable automated component inventory generation, capturing provenance at build time rather than during incident response. Development tooling becomes part of the compliance evidence rather than an additional validation burden.
Long-Term Lifecycle Reality: Why Reproducibility Must Extend Beyond Release
The requirements that apply at certification continue throughout the operational life of the device. A security patch issued years after deployment must be built within a controlled environment consistent with the original release, validated against the same standards, and supported by traceable evidence.
When toolchain configurations are not preserved, updates require reconstruction of environments that may no longer exist in a controlled form. This introduces uncertainty into validation and increases the cost of maintenance over time.
Long-term support strategies treat toolchain configuration as part of the product definition. Frozen toolchain versions maintained across the device lifecycle ensure that builds remain consistent across time. Development infrastructure becomes a controlled component of the system, supporting both maintenance and regulatory compliance.
Enabling Compliance-Ready Development with IAR
Development systems that support continuous validation and reproducibility require toolchains that are designed for integration, traceability, and long-term stability. In practice, this places constraints on how build environments, analysis tools, and security workflows are structured and maintained.
IAR’s embedded development platform is built to operate within these constraints. Its toolchains are TÜV-certified against IEC 62304, contributing directly to the tool qualification component of a medical device compliance case. A unified platform supports Arm, RISC-V, Renesas, and TI MSP430 architectures, reducing the number of separately qualified toolchains across product lines.
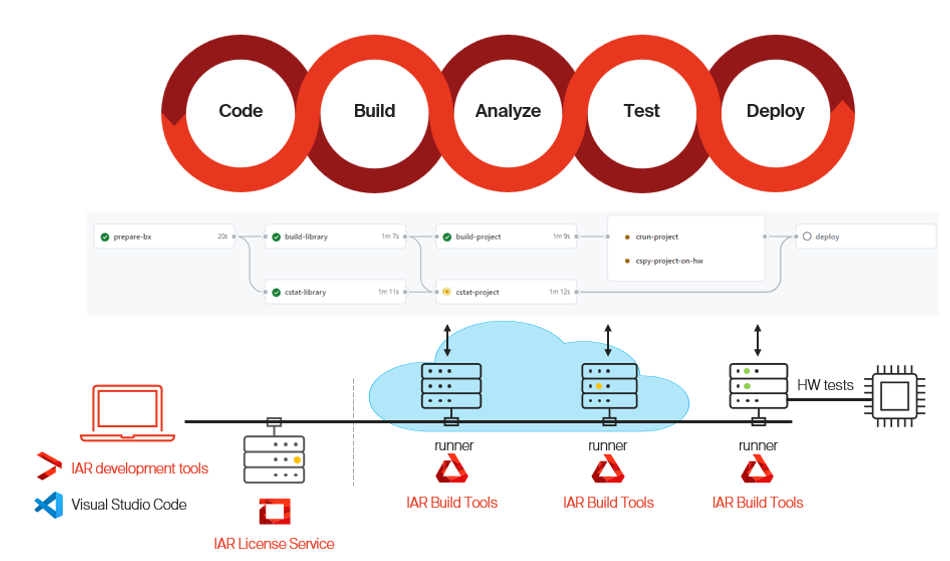
IAR Build Tools are headless and integrate with CI platforms such as GitHub Actions, GitLab CI, and Jenkins. Static analysis through C-STAT enforces MISRA C/C++, CERT C/C++, and CWE guidelines against each build within the CI/CD pipeline, and runtime verification through C-RUN extends coverage into test execution via dynamic instrumentation that detects memory access violations, arithmetic errors, and undefined behavior, enabling continuous validation without manual intervention. Together, C-STAT and C-RUN provide analysis coverage that maps directly to IEC 62304 software verification requirements.
Security capabilities, including firmware signing, encryption, and secure boot through IAR’s Embedded Trust, are integrated into the same workflow, aligning with FDA cybersecurity guidance and CRA requirements.
Long-Term Support versions provide frozen toolchain configurations maintained across extended periods — covering the full operational life of a medical device, commonly 10 to 15 years in active use. Builds produced under a specific configuration can be reproduced years later, with validated service packs and structured issue reporting supporting long-term device maintenance and regulatory requirements. These capabilities align with the structural requirements outlined earlier, where compliance depends on controlled, reproducible development systems rather than isolated validation activities.
From Fragmentation to Lifecycle Control
MedTech development now depends on control of the software lifecycle across its full duration. Fragmented development environments introduce gaps in traceability, increase validation effort, and limit the ability to respond to security events within regulatory timelines.
Continuous, deterministic development processes provide a stable foundation for meeting these requirements. Systems that integrate validation, execution, and compliance into daily workflows reduce lifecycle debt and improve operational predictability. Meeting the 24-hour CRA notification window, maintaining IEC 62304 traceability across a 10–15 year device lifecycle, and delivering validated OTA patches without schedule risk all depend on one thing: compliance evidence that exists before it is needed, not reconstructed under pressure when it is. In MedTech, reproducibility defines the boundary between controlled development and regulatory risk.